Clean Syn-Fuels via Hydrogenation Processes: Acidity–Activity Relationship in O-Xylene Hydrotreating


1
CNR Institute of Advanced Technology for Energy “Nicola Giordano”, 98125 Messina, Italy
2
Institute of Researches on Catalysis and Environment in Lyon (IRCELyon), Lyon, France
3
Department of Engineering, University of Messina, 98158 Messina, Italy
*
Authors to whom correspondence should be addressed.
ChemEngineering 2020, 4(1), 4; https://doi.org/10.3390/chemengineering4010004
Submission received: 24 September 2019
/
Revised: 4 December 2019
/
Accepted: 2 January 2020
/
Published: 6 January 2020
(This article belongs to the Special Issue 2019 HYPOTHESIS XIV)
Abstract
:Transition metal sulfide catalysts are actually the most performing catalytic materials in crude oil hydrotreating (HDT), for energetic purposes. However, these systems suffer from several drawbacks that limit their exploitation. Aiming to meet the even more stringent environmental requirement, through a remarkable improvement of HDT performance in the presence of refractory feedstock (i.e., in terms of activity, selectivity, and stability), a deeper knowledge of the structure–activity relationship of catalysts must be achieved. Therefore, in this study, CoMo/γ-Al2O3 and NiMo/γ-Al2O3 catalysts were characterized and tested in the o-xylene hydrogenation model reaction, assessing the influence of both support acidity and catalyst acid strength on reaction pathway by employing γ-Al2O3 and Y-Type zeolite as acid reference materials. A clear relationship between concentration and strength of acid sites and the performance of the catalytic materials was established. Cobalt based catalyst (CoMoSx) proves a higher acidic character with respect to Nickel (NiMoSx), prompting isomerization reactions preferentially, also reflecting a greater o-xylene conversion. The different chemical properties of metals also affect the catalytic pathway, leading on the CoMoSx system to the preferential formation of p-xylene isomer with respect to m-xylene.
1. Introduction
Due to increased environmental concern, the European Union has introduced the “zero-sulphur” legislation, which limits the sulphur content in gasoline and diesel less than 10 mg/kg [1], thus requiring the development of modern process technologies and the improvement of the actual hydrotreating catalysts for “very real” clean-fuels manufacturing [2,3]. Transition metal sulfide catalysts, usually consisting in molybdenum (Mo) supported on high surface area carriers and promoted by cobalt (Co) or nickel (Ni), are widely used in crude oils hydrotreatment [4,5,6,7,8,9]. The catalytic synergism of these systems relies on the formation of Co(or Ni)MoS phases, where the active sites are placed on both the metal and sulfur edges, and BRIM sites [10,11,12]. The edge sites are sulfur vacancies, so-called coordinatively unsaturated sites (CUS), formed under sulfo-reductive conditions at the edges of MoS2-cluster and stabilized by the presence of promoters [10,11,12,13]. CUS sites exhibit a Lewis acid character and are responsible for the direct hydrogenolysis of C-S, C-N and C-O bond during hydroprocessing [10], while BRIM sites are known to possess metallic characteristics, which allows hydrogenation reactions to occur, thus favoring the hydroconversion of the most refractory compounds [10,12,14,15,16,17].
Then, despite their peculiar activity in removing heteroatoms-containing compounds, these systems suffer from some drawbacks that may limit their application. In particular, catalysts deactivation by coke deposition is one of the major concerns during hydroprocessing, especially of heavy residual crude fractions [18]. It is generally accepted that carbon deposits are formed as a results of polymerization and cracking reactions, involving carbenium ion formation, which take place on acid sites of catalysts [19,20,21,22,23,24,25]; however, the nature of acid sites involved and their role on hydrotreating (HDT) reaction mechanism and deactivation phenomena is still unclear [26,27,28,29]. Indeed, several studies have reported that coke-resistant catalysts generally possess MoSx centers with a low level of Lewis acidity, therefore suggesting that Lewis acid sites participating in HDT and coke-forming reactions are related to the presence of active Mo phases formed during the sulfiding treatment [25]. Conversely, other research claims that the acid character of the HDT catalyst is related to the support only [26,28,30,31]. As proof, the carbon formation can be limited by selecting oxide supports alternative to γ-Al2O3 with lower acidity (such as MgO, ZrO2, TiO2), or modifying the alumina properties through alkali addition (i.e., MgO; B2O3), although the performance results were affected [28,29,32,33,34]. Therefore, the design of new catalytic materials, useful in the conversion of very refractory feedstock, requires a deeper knowledge on both the mechanisms of carbon formation and on the relationships between catalyst structure and activity. In order to improve activity, selectivity, and stability of catalysts even in the presence of refractory feedstock, it is mandatory to assess the role of the active sites of HDT catalysts in carbon formation. On this account, different characterization techniques have been applied. The adsorption of probe molecules followed by a physical technique (ex. Infrared (IR) spectroscopy) is a generally adopted approach for characterizing the acid-base and hydrogenative properties of catalysts [35,36]. However, these techniques are carried out under conditions very different from those of the industrial practice, which makes difficult to translate the information obtained to the real industrial process (i.e., high hydrogen pressure, presence of S-containing compounds) [37]. Then, a more “profitable” way to characterize Co(or Ni)MoS active sites can be the catalytic study of simple model reactions, approaching operating conditions of the industrial process. As documented, the acidic character of solid acid catalysts can be esteemed by isomerization tests, employing organic compound such as xylenes, 1-hexane, n-butane, and pentane [34,37,38,39,40,41,42,43]. In particular, under the conditions of hydroprocessing, xylenes can be hydrogenated through parallel reactions, according to the different families of active sites, reflecting hydrogenation, condensation, polymerization, and isomerization activity [37].
Thus, the present study aims to assess the influence of the catalyst acidity on carbon formation phenomena through the reaction path of hydrogenation of o-xylene. Comparative analysis and catalytic studies have been carried out on γ-Al2O3 supported NiMoSx and CoMoSx catalysts, shedding light on the effect of metals (Co or Ni) promotion on MoSx activity and catalyst selectivity, pointing out the different reaction mechanism of the two metals.
2. Materials and Methods
2.1. Catalytic Materials
Commercialγ-Al2O3 supported nickel-molybdenum (NiMo/γ-Al2O3) and cobalt-molybdenum (CoMo/γ-Al2O3) catalysts, available from Albemarle (Charlotte, NC, USA), were employed for the study. Before tests, the catalysts were ex-situ activated by sulfiding process at 350 °C and 11 bar in flowing H2S/H2 gas mixture (15 vol%) for 1h and coded as CoMoSx and NiMoSx systems. In order to value the effect of support acidity and acid strength on catalyst behavior and reaction path, commercial γ-Al2O3 (Akzo 000-3P) and LaY Zeolite (lanthanum stabilized Y zeolite), available from Grace (Columbia, MD, USA), with a low silicon-to-alumina ratio were used as acid reference materials.
2.2. Catalyst Characterization
Surface area (S.A.), pore volume (P.V.), and average pore diameter (A.P.D.) were determined by nitrogen adsorption–desorption isotherms at −196 °C, employing a Micromeritics ASAP 2020 instrument (Norcross, GA, USA), applying conventional Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halenda (BJH) calculation methods.
X-ray diffraction (XRD) patterns were collected at ambient atmosphere, in the 2θ range 10°–80°, by using a Philips X’Pert powder diffractometer (Amsterdam, Netherlands) operating with Ni β-filtered Cu Kα radiation (40 kV; 30 mA) and a scan rate of 0.05°/min.
Fourier-transform infrared (FTIR) spectra of pre-adsorbed pyridine were recorded by the use of a Bruker FTIR spectrophotometer (Billerica, MA, USA) equipped with a mercury cadmium telluride (MCT) detector, in reflectance mode, using a resolution of 4 cm−1. The samples powder, diluted with KBr (1/1), were activated under vacuum (10−3 mbar) for 1 hour at 150 °C in a DRIFT IR cell allowing in situ thermal treatments, and pyridine dosage at 150 °C under Ar flow (FAr = 30 cc/min).
2.3. Catalytic Study
The o-xylene hydrogenation tests were carried out by using a laboratory semi-automatized microplant equipped with an AISI 316 stain steel up-down Plug Flow Reactor. The tests were performed at 350 °C and 11 bar, feeding 30 stp cc∙min−1 of a reactant mixture of o-xylene (10%), H2 (81.4%), H2S (0.05%), C2H6 (3.6%), and N2 (4.95%); in each test, the reactor was loaded with 1 g of sulfided catalyst diluted one-to-one with quartz pellets. The online composition of reactants and products was determined by using a gas-chromatography (GC) model Agilent 7890A (Santa Clara, CA, USA) equipped with thermo-conductivity (TCD) and flame ionization (FID) detectors. Permanent gases (H2, N2, C2H6) were detected by using a Molecular Sieves and a Porapak Q packed columns connected in series. For GC separation of condensable hydrocarbons, a Supelcowax 10 fused silica capillary column (30 m × 0.53 mm ID, 2 μm film, Sigma-Aldrich (St. Louis, MO, USA)) coated with polar polyethylene glycol stationary phase was used. The column temperature was held at 40 °C for 5 min and then programmed at 5 °C·min−1 at 160 °C, using helium as carrier gas. In these conditions, a good separation of all reaction products, including cis-/trans-dimethylcyclohexanes stereoisomers was obtained. Then, the response factors of each compound were determined for the calibration of the GC signals by using several standards. Catalytic results were reported in terms of o-xylene conversion (i.e., % Xo-xyl), selectivity to different products (i.e., % Spi) and specific reaction rate (i.e., mol∙gcat−1∙h−1), (Equations (1)–(3)):
3. Results and Discussion
3.1. Characterization of Catalysts
The list of catalysts with the main chemical-physical properties and acidity is reported in Table 1. The catalysts were investigated as CoMo and NiMo oxides and sulfides, quenching and storing into inert gas the sulfides sample in order to prevent and avoid any re-oxidation phenomena.
According to the different chemical composition, the samples show variable total surface area, ranging from about 100 m2/g (NiMoSx) to 450 m2/g (LaY), also mirroring similar acidity scale, obtained through FTIR measurements, as in more detail discussed in the next paragraphs.
As shown in Figure 1a, the open channel structure of the Y-type zeolite (i.e., Faujasite crystal morphology) confirms the XRD signals, while the XRD pattern of γ-Al2O3 refers also to the presence of isolated amorphous Al2O3 phases [44]—then, a part of the XRD reflections that confirms the presence of γ-Al2O3 support, the absence of any diffraction lines belonging to the crystal structure of cobalt-molybdenum sulfide that accounts for highly dispersed cobalt-molybdenum precursors [45]. Then, among the diffraction peaks of support, the XRD pattern of NiMoSx catalyst proves the formation of NiSO4 species, with XRD signals at 2-theta degree of 20°, due to Ni segregation phenomena.
Then, FTIR measurements were carried out after catalyst exposure to pyridine (as probe molecule) in order to evaluate the acidic properties of catalysts, in terms of availability, strength and nature of acid sites, Figure 1b. Then, according to the peaks’ intensity, NiMoSx catalyst results with much less acid than CoMoSx. Namely, all samples feature the ring-stretching mode corresponding to pyridine bonded with Brønsted (B, i.e., 1545 cm−1; 8a mode) and Lewis (L, i.e., 1446 cm−1; 19b mode) sites, respectively, resulting in the band at 1490 cm−1 (19a mode) from the contribution of both Lewis and Brønsted sites [46,47]. The concentration of Lewis and Brønsted acid sites (CLA and CBA, respectively) on the catalyst surface was esteemed by peaks’ integration, as summarized in Table 1. In particular, CoMoSx catalyst records modest acid properties, with a strong predominance of Lewis sites, estimated as 0.380 mmol·gcat−1. Instead, the NiMoSx catalyst exhibits a poor acidic character, resulting of 0.117 and 0.032 mmol·gcat−1 the availability of Lewis and Brønsted sites, respectively. Thus, γ-Al2O3 reference sample shows a concentration of acid sites quantified in circa 0.330 mmol·gcat−1 with the lowest presence of Brønsted sites with the respect to the other samples, Table 1 and Figure 1b. The system at highest acidity, LaY-zeolite, also reveals a greater contribution of Lewis sites, reporting a surface concentration of acid sites of 1.531 mmol·gcat−1 and 0.968 mmol·gcat−1, due to Lewis or Brønsted sites, respectively.
3.2. Catalytic Study
In Figure 2, the reaction scheme of o-xylene hydrogenation process is reported, according to products detected, which namely account for reactions of direct hydrogenation (cis-/trans-1,2-dimethylcyclohexane), isomerization and isomerization-hydrogenation (p-xylene, m-xylene, cis-/trans-1,3-dimethylcyclohexane and cis-/trans-1,4-dimethylcyclohexane), disproportion (toluene and trimethylbenzene), and polycondensation-cracking phenomena (other species).
The results of the catalytic measurements are summarized in Table 2, in terms of o-xylene conversion, ratio between p-xylene and m-xylene selectivity (i.e. p-/m-) and specific reaction rates (i.e., isomerization (ISOM), hydrogenation (HYD), disproportion (DIS), and polycondensation (POL) rates), while, in the graphics of Figure 3, the catalytic behaviors of the different samples as a function of time-of-stream (t.o.s.) are compared.
As shown in Table 2, the catalytic conversion of o-xylene perfectly reflects the catalysts scale of activity:
LaY > CoMoSx > NiMoSx > γ-Al2O3.
In particular, γ-Al2O3 signs a very poor catalytic activity in comparison to all catalysts, with a global conversion of o-xylene of 0.37% achieved in 24 h, accounting for only “non-hydrogenated” products (i.e., isomers, disproportion, and polycondensation). In spite of the greater catalytic activity, with a conversion at 24 h of 47.4%, the LaY system does not promote the hydrogenation reactions, leading to the larger carbon deposition via polycondensation reactions (Table 2) similarly to the γ-Al2O3 sample. This proves of an acid-basic catalytic behavior, also prefiguring the role and the mechanism carried out by acid sites in the o-xylene hydrotreating processes. Diversely, the hydrogenation functionality is evident on both CoMoSx and NiMoSx catalytic systems. A similar catalytic behavior was evidenced by Kaluža in the isomerization of 1-chyclohexene under hydrotreating condition over sulfided transition metals (Co, Mo, Ni, W) and noble metals (Rh, Pd, Ir, Pt, Re) catalysts [33]. In this study, in fact, the author observed that the isomerization path proceeded very fast over all catalysts and supports, although, like what was observed for the o-xylene transformation, only sulfided transition metals were able to catalyze the hydrogenation of methylcyclohexenes [33].
Then, the catalytic results here reported also attest to the greater activity of Co-based catalyst which achieves at 24 h a conversion of o-xylene higher than 5.00%, although the direct hydrogenation of o-xylene is less promoted with respect to the isomerization reactions. At the steady-state condition, NiMoSx catalyst reports ca. 2% of conversion with an enhanced activity for the hydrogenation reactions (Table 2), pointing out that the properties of metal affect the reaction path.
Irrespectively to structure and chemical composition, all samples report a rapid loss of activity in the early 10 h of reaction, which is even more evident on the most active catalysts (i.e., LaY and CoMoSx), where the conversion falls down in less than 24 h of ca. 60% (Figure 3A). The observed decrease of activity along the first hours of reaction has as a main consequence a progressive decrease of selectivity versus the products of polycondensation. These findings agree with the occurring of carbon deposition processes on the catalyst surface. As known, many hydrotreating process see a huge amount of carbon deposited on the surface within the first 3–24 h [21,23]. In the industrial practice, it was well established that, after the initial rapid coke build-up, the coking process slows down and tends to level off or reach an equilibrium value within 24 h. On this account, we observe a fast and progressive deactivation of the catalyst, highlighted by the remarkable drop of conversion in the first 3–10 h (Figure 3), which reflects the catalyst acidity on the surface. Indeed, as a consequence of the highest concentration of acid sites (Table 1), LaY and CoMoSx samples are mainly affected by the carbon build-up process, which drives to faster deactivation. Accordantly, carbon deposition is remarkably lower on the less acid NiMoSx catalyst, in fact, reaching the steady-state condition in only 5 h, the Ni-based system shows a selectivity to “coke” of about 9% signing a highly stable behavior (Figure 3). Then, the contribution of the support acidity to the catalytic activity of CoMoSx and NiMoSx systems is clearly deducted from the results of catalytic test with the reference γ-Al2O3. In particular, the γ-Al2O3 sample shows very poor activity, promoting exclusively the reaction of cracking, poly-condensation, and polymerization, due to the acid properties only.
3.3. Acidity–Activity Relationships
Figure 4 details on the product distribution obtained with the different catalysts. As shown, due to the impossibility of activating hydrogen, LaY and γ-Al2O3 reference materials catalyze exclusively the reactions of isomerization and disproportion, which lead to p-xylene, m-xylene, toluene, and tri-methylbenzene as products of o-xylene conversion. In particular, γ-Al2O3 catalyzes majorly the isomerization to m-xylene with respect to p-xylene, reporting a p-/m- ratio of 0.17 (Table 1). Diversely, the selectivity to 1,4- isomer results remarkably favored on LaY sample, attesting p-/m- ratio of 3.55, Table 1.
The formation of various saturated napthenes, due to the hydrogenative functionality, makes the catalytic pathway of CoMSx and NiMoSx systems more complex, Figure 4. In particular, the NiMoSx sample prompts preferentially the direct hydrogenation of o-xylene, with a selectivity to cis-/trans-1,2-dimethylcyclohexanes of about 50%. In the opposite way, the CoMoSx catalyst drives more preferentially to isomers and hydrogenated isomers of o-xylene. In addition, on a nickel based catalyst, m-xylene is the more preferred isomer, with a p-/m- ratio of 0.44, while CoMoSx promotes mainly the formation of 1,4-isomers, with p-/m- ratio of 3.10, Table 2.
As reported in literature, isomerization of di-alkylaromatics by acid materials can occur with three pathways, which are: (i) intramolecular, proceeding through methyl shifts in benzenium ion intermediates, (ii) intermolecular, involving trans-alkylation, and (iii) dissociative by reversible de-alkylation–alkylation of tert-alkyl-aromatics [37,41,42].
Accordingly to the mechanism proposed by Wilson et al., m-xylene is the initial product of a methyl-shift, where the low p-/m-ratio reported by γ-Al2O3 and NiMoSx systems is consistent with the unimolecular isomerization pathway [41]. In contrast, the higher p-/m- ratio recorded with LaY and CoMoSx samples can be justified by a different intermolecular isomerization pathway or by a stronger intermediate adsorption that prevents the desorption of m-xylene before further methyl-shift reaction. Finally, the selectivity towards other by-products, deriving from polycondensation and cracking reactions, deserves special attention. In fact, not following any relationship with the catalytic activity in the o-xylene transformation, POL reactions are mainly favored on the γ-Al2O3 sample, which shows a selectivity to the side products higher than 50%, pointing out that a more complex mechanism may rule the occurrence of polycondensation/cracking reactions.
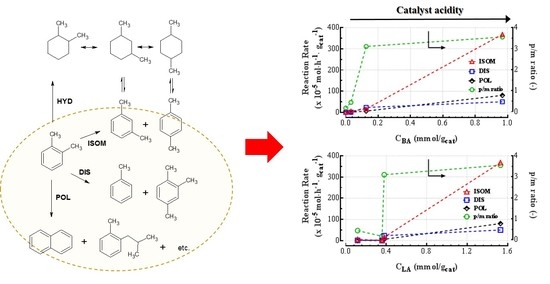
Therefore, the activity for both the isomerization and disproportion of o-xylene can be considered as representative of the acid activity of the catalysts. Moreover, the p-/m- ratio and the polycondensation/cracking reactions may also be affected by the acid strength of the system’s surface. On this account, Figure 5 correlates the catalytic data of the different materials studied with the concentration of Brønsted and Lewis acid sites determined by FTIR measurements.
In particular, an almost linear relationship between ISOM and DIS reaction rates and the concentration of both Brønsted acid sites can be evidenced. Indeed, the participation of Brønsted acid sites in the xylenes isomerization reaction has been clearly demonstrated by correlations between the isomerization rate and the concentration of protonic sites of different solids, such as silica, zeolites, zirconia, etc. [41,43,48,49]. However, the increasing ISOM and DIS rates with the increasing of Lewis-type acidity may indicate that Lewis acid sites can also take part to the o-xylene isomerization through a radical cation mechanism [41,48]. Nonetheless, this pathway seems to be less favored, as proved by the very low activity obtained with the γ-Al2O3 sample, in which only Lewis-type acid sites are present. Then, the increase of p-/m- ratio with the concentration of Brønsted acid sites further demonstrates the occurrence of the intermolecular methyl-shift pathway for xylene isomerization reactions. In fact, a stronger acidity of the surface means a stronger adsorption of electron-donating molecules; thus, m-xylene desorption is hindered, favoring the subsequent isomerization to p-xylene. Consequently, Lewis acid sites seem to be in larger part responsible for polycondensation-cracking reactions, as proved by the direct proportionality between POL rate and CLA. Polycondensation reactions lead to the coupling of several polyaromatic entities in the feedstock which than give rise to more polar structures of progressively increasing molecular weight. These species agglomerate to form a film of variable thickness on the surface of the catalyst, provoking the catalyst deactivation by coking [49]. Therefore, these results corroborate that Lewis acidity is one of the key factor in deactivation phenomena of HDT catalysts. Likewise, considering the poorer performance of γ-Al2O3 sample, the strongest CUS sites at the edges of Co(Ni)MoSx slabs, which possess an high Lewis-type acidity, may be the active centers involved in the polycondensation as well as in the o-xylene isomerization reactions [25].
In addition, both distribution and strength of acid sites may vary considerably under the process conditions of hydrotreating, due to possible interactions between the Brønsted and Lewis acid centers with H2 molecules. On this account, in Table 3 are summarized the catalytic data obtained performing the o-xylene HDT process with Co(Ni)MoSx systems under inert atmosphere.
Without hydrogen, both CoMoSx and NiMoSx catalysts display a very low activity, with similar o-xylene conversion values lower than 1%. Moreover, the only reaction occurring is the direct isomerization to m- and p-xylenes, with a p-/m- ratio of 0.23 for both systems. The poorer performance obtained with the inert with respect to that recorded under hydrogen pressure, clearly demonstrate that the xylene isomerization pathway is strongly influenced by the interaction between H2 and the catalysts active sites, even though hydrogen is not directly involved in the reaction stoichiometry and H2 transfer steps are not required for the isomerization reaction [41]. Moreover, DIS reaction are completely hindered in the absence of hydrogen. Since disproportion reactions are known to occur through a bimolecular mechanism, needing two adjacent active sites, the presence of hydrogen influences not only the strength of the acid site (as proved by the higher p-/m- ratio) but also the density of the Brønsted sites on the catalyst surface. On this account, as reported by Wilson et al. for WO-based catalysts, Lewis acid sites may also act as Brønsted acidity precursors under hydrogenative conditions; H atoms can interact with TM6+ entities with a Lewis character, producing active Brønsted sites, thus increasing their surface density and strength [41].
4. Conclusions
The hydrotreating of an o-xylene model compound effectively allows for measuring the hydrogenating activity and the acid activity of HDT catalysts under industrial conditions (high hydrogen pressure, presence of S- containing compounds). In particular, the hydrogenating activity can be characterized by the activity for the transformation of o-xylene into dimethylcyclohexanes, while the isomerization and disproportion path can be considered as representative of the acid activity of the materials. As proof, the catalytic performance of typical Co(Ni)MoSx catalysts have been compared with those recorded with γ-Al2O3 and La-stabilized Y-type zeolite as reference materials of low and high acidity, respectively. Namely, an almost linear relationship between ISOM and DIS reaction rates and the concentration of both Brønsted and Lewis acid sites, estimated through FTIR spectroscopic measurements, can be evidenced. Moreover, the presence of cobalt or nickel deeply influences both chemical-physical and catalytic properties of HDT catalysts. In particular, the CoMoSx catalyst is characterized by a stronger acidic character than the NiMoSx one. As a result, the CoMoSx catalyst shows a better catalytic performance in the isomerization/hydrogenation reaction, catalyzing preferentially the isomerization of o-xylene rather than the hydrogenation route. However, the higher the catalyst surface Lewis-type acidity, the faster the catalyst coking is, as evidenced by the direct proportionality between the rates of polycondensation reactions (POL) and the concentration of Lewis acid sites.
Author Contributions
Conceptualization, L.S. and A.P.; methodology, L.S, A.P., K.B., and F.A.; investigation, A.P. and K.B.; writing—original draft preparation, A.P. and K.B.; writing—review and editing, L.S. and F.A.; supervision, L.S. and F.A.; project administration, L.S.; funding acquisition, L.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Eco-Rigen S.r.l. is gratefully acknowledged for the financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- CEC DIRECTIVE 2009/30/EC of the European Parliament and of the Council. Off. J. Eur. Union 2009, 136–148. [CrossRef]
- Guo, R.; Cao, Z.; Fang, X. The development of catalysts and their stacking technology for diesel ultra-deep hydrosulfurization. Catal. Today 2018, 316, 21–25. [Google Scholar] [CrossRef]
- Pereyma, V.Y.; Klimov, O.V.; Prosvirin, I.P.; Gerasimov, E.Y.; Yashnik, S.A.; Noskov, A.S. Effect of thermal treatment on morphology and catalytic performance of NiW/Al2O3 catalysts prepared using citric acid as chelating agent. Catal. Today 2018, 305, 162–170. [Google Scholar] [CrossRef]
- Nikulshin, P.A.; Mozhaev, A.V.; Maslakov, K.I.; Pimerzin, A.A.; Kogan, V.M. Genesis of HDT catalysts prepared with the use of Co2Mo10HPA and cobalt citrate: Study of their gas and liquid phase sulfidation. Appl. Catal. B Environ. 2014, 158–159, 161–174. [Google Scholar] [CrossRef]
- Nikulshin, P.A.; Salnikov, V.A.; Mozhaev, A.V.; Minaev, P.P.; Kogan, V.M.; Pimerzin, A.A. Relationship between active phase morphology and catalytic properties of the carbon-alumina-supported Co(Ni)Mo catalysts in HDS and HYD reactions. J. Catal. 2014, 309, 386–396. [Google Scholar] [CrossRef]
- Spadaro, L.; Palella, A.; Frusteri, F.; Arena, F. Valorization of crude bio-oil to sustainable energy vector for applications in cars powering and on-board reformers via catalytic hydrogenation. Int. J. Hydrogen Energy 2015, 40, 14507–14518. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Nishi, M.; Mochizuki, T.; Takagi, H.; Takatsuki, A.; Roschat, W.; Toba, M.; Yoshimura, Y. Co-processing of Jatropha-derived bio-oil with petroleum distillates over Mesoporous CoMo and NiMo Sulfide Catalysts. Catalysts 2018, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Díaz de León, J.; Ramesh Kumar, C.; Antúnez-García, J.; Fuentes-Moyado, S. Recent Insights in Transition Metal Sulfide Hydrodesulfurization Catalysts for the Production of Ultra Low Sulfur Diesel: A Short Review. Catalysts 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhu, H.; Zhao, J.; Pan, L.; Dai, P.; Gu, X.; Li, L.; Liu, Y.; Zhao, X. Synthesis of Mesoporous γ-Al2O3 with Spongy Structure: In-Situ Conversion of Metal-Organic Frameworks and Improved Performance as Catalyst Support in Hydrodesulfurization. Materials 2018, 11, 1067. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Garrido, I.; López-Benítez, A.; Berhault, G.; Guevara-Lara, A. Effect of support on the acidity of NiMo/Al2O3-MgO and NiMo/TiO2-Al2O3 catalysts and on the resulting competitive hydrodesulfurization/hydrodenitrogenation reactions. Fuel 2019, 236, 55–64. [Google Scholar] [CrossRef]
- Besenbacher, F.; Brorson, M.; Clausen, B.S.; Helveg, S.; Hinnemann, B.; Kibsgaard, J.; Lauritsen, J.V.; Moses, P.G.; Nørskov, J.K.; Topsøe, H. Recent STM, DFT and HAADF-STEM studies of sulfide-based hydrotreating catalysts: Insight into mechanistic, structural and particle size effects. Catal. Today 2008, 130, 86–96. [Google Scholar] [CrossRef]
- Lauritsen, J.V.; Bollinger, M.V.; Lægsgaard, E.; Jacobsen, K.W.; Nørskov, J.K.; Clausen, B.S.; Topsøe, H.; Besenbacher, F. Atomic-scale insight into structure and morphology changes of MoS2 nanoclusters in hydrotreating catalysts. J. Catal. 2004, 221, 510–522. [Google Scholar] [CrossRef]
- Breysse, M.; Geantet, C.; Afanasiev, P.; Blanchard, J.; Vrinat, M. Recent studies on the preparation, activation and design of active phases and supports of hydrotreating catalysts. Catal. Today 2008, 130, 3–13. [Google Scholar] [CrossRef]
- Topsøe, H. The role of Co-Mo-S type structures in hydrotreating catalysts. Appl. Catal. A Gen. 2007, 322, 3–8. [Google Scholar] [CrossRef]
- Lauritsen, J.V.; Nyberg, M.; Nørskov, J.K.; Clausen, B.S.; Topsøe, H.; Lægsgaard, E.; Besenbacher, F. Hydrodesulfurization reaction pathways on MoS2 nanoclusters revealed by scanning tunneling microscopy. J. Catal. 2004, 224, 94–106. [Google Scholar] [CrossRef]
- Li, J.; Zhang, M.; Guan, Z.; Li, Q.; He, C.; Yang, J. Synergistic effect of surface and bulk single-electron-trapped oxygen vacancy of TiO2 in the photocatalytic reduction of CO2. Appl. Catal. B Environ. 2017, 206, 300–307. [Google Scholar] [CrossRef]
- Nan, F.; Song, C.; Zhang, J.; Hui, R.; Chen, J.; Fairbridge, C.; Botton, G.A. STEM HAADF Tomography of Molybdenum Disulfide with Mesoporous Structure. ChemCatChem 2011, 3, 999–1003. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.K.; Ray, S.S.; Singh, I.D. Structural characterization of coke on spent hydroprocessing catalysts used for processing of vacuum gas oils. Appl. Catal. A Gen. 2004, 278, 83–91. [Google Scholar] [CrossRef]
- Furimsky, E.; Massoth, F.E. Deactivation of hydroprocessing catalysts. Catal. Today 1999, 52, 381–495. [Google Scholar] [CrossRef]
- Marafi, M.; Stanislaus, A. Influence of catalyst acidity and feedstock quality on hydrotreating catalyst deactivation by coke deposition. Pet. Sci. Technol. 2001, 19, 697–710. [Google Scholar] [CrossRef]
- Marafi, M.; Stanislaus, A. Effect of initial coking on hydrotreating catalyst functionalities and properties. Appl. Catal. A Gen. 1997, 159, 259–267. [Google Scholar] [CrossRef]
- Wiwel, P.; Zeuthen, P.; Jacobsen, A.C. Initial coking and deactivation of hydrotreating catalysts by real feeds. Stud. Surf. Sci. Catal. 1991, 68, 257–264. [Google Scholar] [CrossRef]
- Maity, S.K.; Blanco, E.; Ancheyta, J.; Alonso, F.; Fukuyama, H. Early stage deactivation of heavy crude oil hydroprocessing catalysts. Fuel 2012, 100, 17–23. [Google Scholar] [CrossRef]
- Lebreton, R.; Brunet, S.; Pérot, G.; Harlé, V.; Kasztelan, S. Deactivation and characterization of hydrotreating NiMo/Al2O3 catalyst coked by anthracene. In Catalyst Deactivation; Delmon, B., Froment, G.F., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 195–201. [Google Scholar]
- Scaroni, A.W.; Jenkins, R.G.; Utrilla, J.R.; Walker, P.L. Lewis acidity and coking of hydrodesulfuration catalysts. Fuel Process. Technol. 1984, 9, 103–108. [Google Scholar] [CrossRef]
- Lewandowski, M.; Sarbak, Z. Hydrofining activity and acid-base properties of nickel-molybdenum catalysts incorporated on sodium and magnesium ions-modified alumina. Appl. Catal. A Gen. 1998, 168, 179–185. [Google Scholar] [CrossRef]
- Marques, J.; Guillaume, D.; Merdrignac, I.; Espinat, D.; Brunet, S. Effect of catalysts acidity on residues hydrotreatment. Appl. Catal. B Environ. 2011, 101, 727–737. [Google Scholar] [CrossRef]
- Leyva, C.; Ancheyta, J.; Travert, A.; Maugé, F.; Mariey, L.; Ramírez, J.; Rana, M.S. Activity and surface properties of NiMo/SiO2-Al2O3 catalysts for hydroprocessing of heavy oils. Appl. Catal. A Gen. 2012, 425–426, 1–12. [Google Scholar] [CrossRef]
- Rana, M.S.; Huidobro, M.L.; Ancheyta, J.; Gómez, M.T. Effect of support composition on hydrogenolysis of thiophene and Maya crude. Catal. Today 2005, 107, 346–354. [Google Scholar] [CrossRef]
- Chen, W.; Maugé, F.; Van Gestel, J.; Nie, H.; Li, D.; Long, X. Effect of modification of the alumina acidity on the properties of supported Mo and CoMo sulfide catalysts. J. Catal. 2013, 304, 47–62. [Google Scholar] [CrossRef]
- Rana, M.S.; Ancheyta, J.; Maity, S.K.; Rayo, P. Hydrotreating of maya crude oil: I. Effect of support composition and its pore-diameter on asphaltene conversion. Pet. Sci. Technol. 2007, 25, 187–199. [Google Scholar] [CrossRef]
- Kaluža, L.; Gulková, D.; Vít, Z.; Zdražil, M. High-activity MgO-supported CoMo hydrodesulfurization catalysts prepared by non-aqueous impregnation. Appl. Catal. B Environ. 2015, 162, 430–436. [Google Scholar] [CrossRef]
- Kaluža, L. Activity of transition metal sulfides supported on Al2O3, TiO2 and ZrO2 in the parallel hydrodesulfurization of 1-benzothiophene and hydrogenation of 1-methyl-cyclohex-1-ene. React. Kinet. Mech. Catal. 2015, 114, 781–794. [Google Scholar] [CrossRef]
- Giraldo, S.A.; Centeno, A. Isomerization and cracking under HDS conditions using γ-alumina modified with boron as catalysts support. Catal. Today 2008, 133, 255–260. [Google Scholar] [CrossRef]
- Huo, Q.; Dou, T.; Zhao, Z.; Pan, H. Synthesis and application of a novel mesoporous zeolite L in the catalyst for the HDS of FCC gasoline. Appl. Catal. A Gen. 2010, 381, 101–108. [Google Scholar] [CrossRef]
- Pérez-Martínez, D.J.; Eloy, P.; Gaigneaux, E.M.; Giraldo, S.A.; Centeno, A. Study of the selectivity in FCC naphtha hydrotreating by modifying the acid-base balance of CoMo/γ-Al2O3 catalysts. Appl. Catal. A Gen. 2010, 390, 59–70. [Google Scholar] [CrossRef]
- Guisnet, M.; Thomazeau, C.; Lemberton, J.L.; Mignard, S. Model reaction for the inSitu Characterization of the hydrogenative and acid properties of Industrial Hydrocracking Catalysts. J. Catal. 1995, 151, 102–110. [Google Scholar] [CrossRef]
- Watanabe, K.; Oshio, N.; Kawakami, T.; Kimura, T. Isomerization reactions with sulfur-containing pentane over Metal/SO42−/ZrO2 catalysts. Appl. Catal. A Gen. 2004, 272, 281–287. [Google Scholar] [CrossRef]
- Shi, G.; Fang, D.; Shen, J. Hydroisomerization of model FCC naphtha over sulfided Co(Ni)-Mo(W)/MCM-41 catalysts. Microporous Mesoporous Mater. 2009, 120, 339–345. [Google Scholar] [CrossRef]
- Karim, A.H.; Triwahyono, S.; Jalil, A.A.; Hattori, H. WO3 monolayer loaded on ZrO2: Property–activity relationship in n-butane isomerization evidenced by hydrogen adsorption and IR studies. Appl. Catal. A Gen. 2012, 433, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.D.; Barton, D.G.; Baertsch, C.D.; Iglesia, E. Reaction and Deactivation Pathways in Xylene Isomerization on Zirconia Modified by Tungsten Oxide. J. Catal. 2000, 194, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Zuo, D.; Li, D.; Nie, H.; Shi, Y.; Lacroix, M.; Vrinat, M. Acid-base properties of NiW/Al2O3 sulfided catalysts: Relationship with hydrogenation, isomerization and hydrodesulfurization reactions. J. Mol. Catal. A Chem. 2004, 211, 179–189. [Google Scholar] [CrossRef]
- Guisnet, M.; Gnep, N.S.; Morin, S. Mechanisms of xylene isomerization over acidic solid catalysts. Microporous Mesoporous Mater. 2000, 35, 47–59. [Google Scholar] [CrossRef]
- Spadaro, L.; Arena, F.; Di Chio, R.; Palella, A. Definitive Assessment of the Level of Risk of Exhausted Catalysts: Characterization of Ni and V Contaminates at the Limit of Detection. Top. Catal. 2019, 62, 266–272. [Google Scholar] [CrossRef]
- Liu, B.; Liu, L.; Chai, Y.; Zhao, J.; Li, Y.; Liu, D.; Liu, Y.; Liu, C. Effect of sulfiding conditions on the hydrodesulfurization performance of the ex-situ presulfided CoMoS/γ-Al2O3 catalysts. Fuel 2018, 234, 1144–1153. [Google Scholar] [CrossRef]
- Penkova, A.; Bobadilla, L.F.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. Pyridine adsorption on NiSn/MgO–Al2O3: An FTIR spectroscopic study of surface acidity. Appl. Surf. Sci. 2014, 317, 241–251. [Google Scholar] [CrossRef]
- Mochizuki, T.; Itou, H.; Toba, M.; Miki, Y.; Yoshimura, Y. Effects of Acidic Properties on the Catalytic Performance of CoMo Sulfide Catalysts in Selective Hydrodesulfurization of Gasoline Fractions. Energy Fuels 2008, 22, 1456–1462. [Google Scholar] [CrossRef]
- Franklin, J.L.; Nicholson, D.E. A Kinetic Study of the Decomposition of Hydrocarbons by Silica–Alumina Catalysts. J. Phys. Chem. 1956, 60, 59–62. [Google Scholar] [CrossRef]
- Guichard, B. Life Cycle of an HDT Catalyst. In Catalysis by Transition Metal Sulfides; Thoulhoat, H., Raybaud, P., Eds.; Editions Technip: Paris, France, 2013; pp. 301–357. ISBN 9782710809913. [Google Scholar]
Figure 1.
(a) X-ray diffraction patterns and (b) Fourier-transform infrared (FTIR) spectra of adsorbed pyridine of catalysts.
Figure 1.
(a) X-ray diffraction patterns and (b) Fourier-transform infrared (FTIR) spectra of adsorbed pyridine of catalysts.

Figure 2.
Reaction network of o-xylene hydrotreating (HDT).

Figure 3.
Catalytic results: (A) conversion of o-xylene and (B) selectivity to polymerization and cracking products.
Figure 3.
Catalytic results: (A) conversion of o-xylene and (B) selectivity to polymerization and cracking products.

Figure 4.
Product distribution in the o-xylene HDT process recorded at 24 h of t.o.s.

Figure 5.
Correlation between catalytic data and the concentration of Brønsted and Lewis acid sites determined by FTIR measurements.
Figure 5.
Correlation between catalytic data and the concentration of Brønsted and Lewis acid sites determined by FTIR measurements.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Chemical-physical properties and acid sites quantification of catalysts.
| Catalyst | Composition (wt%) | S.A.BET (m2/g) | A.P.D. (nm) | P.V. (cm3/g) | CLA * (mmol/gcat) | CBA ** (mmol/gcat) | |||
|---|---|---|---|---|---|---|---|---|---|
| CoS or NiS | MoS2 | Al2O3 | SiO2 | ||||||
| CoMoSx | 7.29 | 27.5 | 65.2 | - | 215 | 8.0 | 0.33 | 0.380 | 0.126 |
| NiMoSx | 14.0 | 10.0 | 76.0 | - | 102 | 9.1 | 0.28 | 0.117 | 0.032 |
| LaY | - | - | 56.5 | 38.6 | 450 | 2.5 | 0.30 | 1.531 | 0.968 |
| -Al2O3 | - | - | 100 | - | 242 | 8.8 | 0.50 | 0.330 | 0.029 |
* esteemed by FTIR spectra (integration of peak at 1446 cm−1); ** esteemed by FTIR spectra (integration of peak at 1545 cm−1). S.A.—Surface area, P.V.—pore volume, A.P.D.—average pore diameter, CLA, CBA—the concentration of Lewis and Brønsted acid sites, respectively.
Table 2.
Catalytic activity data at 24 h of t.o.s.
| Catalyst | Χo-xylene (%) | Reaction Rates (×10−5 mol·h−1∙gcat−1) | p-/m- | |||
|---|---|---|---|---|---|---|
| ISOM | HYD | DIS | POL | |||
| LaY | 47.4 | 368 | 0.00 | 50.3 | 80.7 | 3.55 |
| CoMoSx | 5.33 | 16.3 | 15.8 | 23.4 | 6.50 | 3.10 |
| NiMoSx | 1.81 | 6.26 | 11.4 | 1.22 | 1.67 | 0.44 |
| γ-Al2O3 | 0.37 | 1.73 | 0.00 | 0.19 | 1.88 | 0.17 |
Table 3.
Catalytic activity data at 24 h of t.o.s. under N2 atmosphere.
| Catalyst | Χo-xylene (%) | Reaction Rates (×10−5 mol·h−1∙gcat−1) | p-/m- | ||
|---|---|---|---|---|---|
| ISOM | HYD | DIS | |||
| CoMoSx | 0.79 | 2.32 | 0.00 | 0.00 | 0.23 |
| NiMoSx | 0.60 | 2.29 | 0.00 | 0.00 | 0.23 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Palella, A.; Barbera, K.; Arena, F.; Spadaro, L. Clean Syn-Fuels via Hydrogenation Processes: Acidity–Activity Relationship in O-Xylene Hydrotreating. ChemEngineering 2020, 4, 4. https://doi.org/10.3390/chemengineering4010004
AMA Style
Palella A, Barbera K, Arena F, Spadaro L. Clean Syn-Fuels via Hydrogenation Processes: Acidity–Activity Relationship in O-Xylene Hydrotreating. ChemEngineering. 2020; 4(1):4. https://doi.org/10.3390/chemengineering4010004
Chicago/Turabian StylePalella, Alessandra, Katia Barbera, Francesco Arena, and Lorenzo Spadaro. 2020. "Clean Syn-Fuels via Hydrogenation Processes: Acidity–Activity Relationship in O-Xylene Hydrotreating" ChemEngineering 4, no. 1: 4. https://doi.org/10.3390/chemengineering4010004